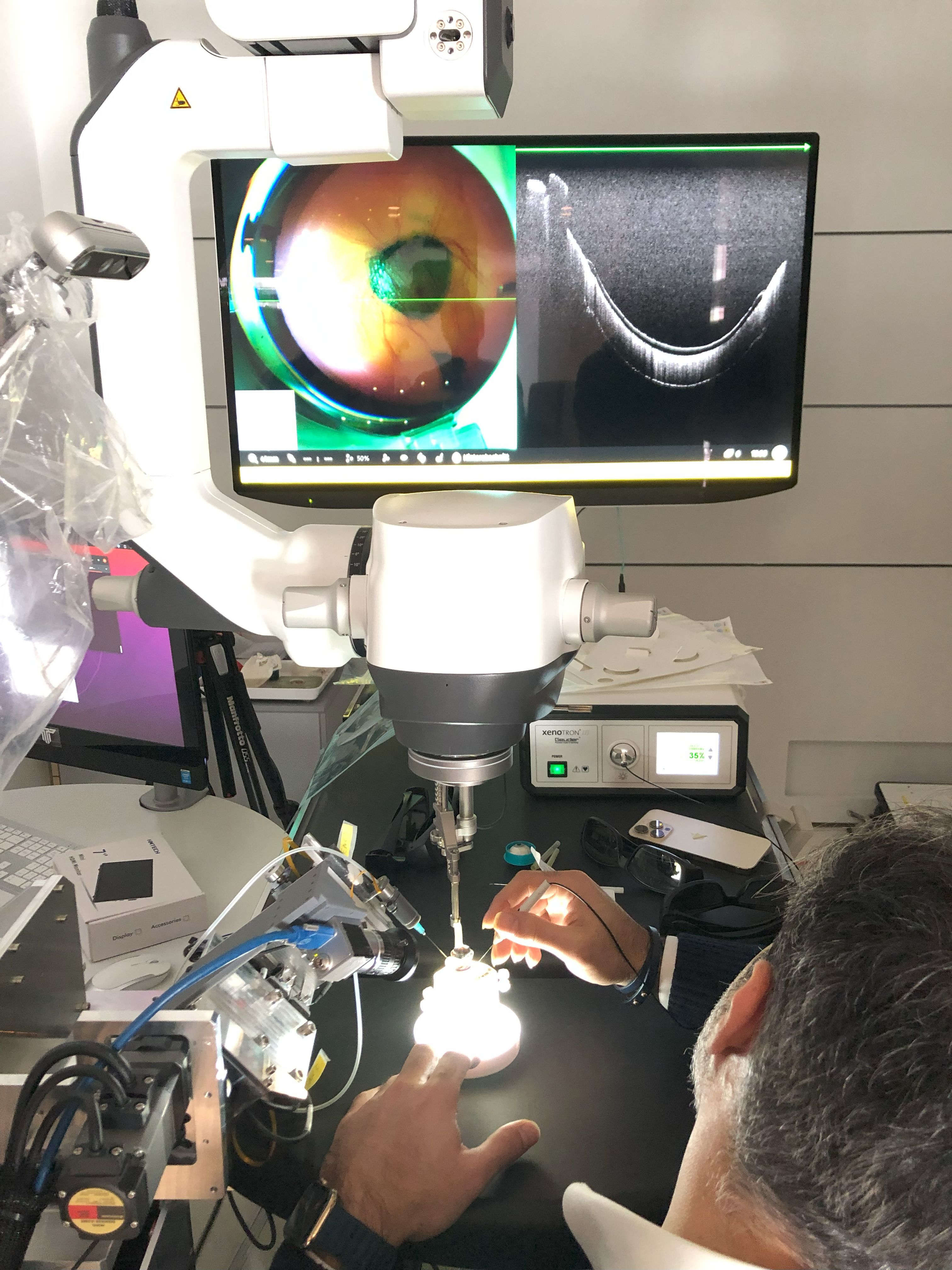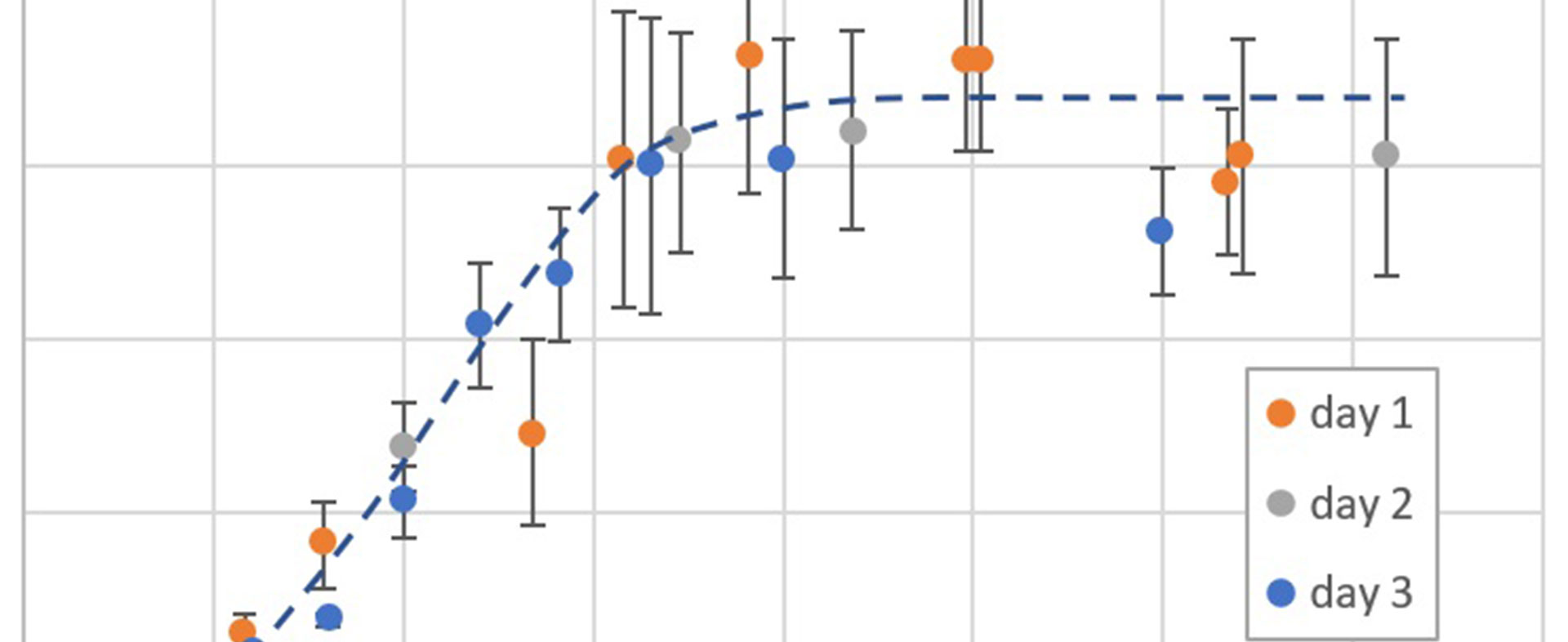



Ziel des Forschungsprojekts war die Entwicklung von anti-herpesviralen Breitbandinhibitoren mit reduzierter Resistenzentwicklung.
Virale Enzyme stellen die wesentlichen Zielmoleküle heute eingesetzter antiviraler Medikamente dar. Diese Arzneien weisen eine Reihe von Nachteilen auf (Nebenwirkungen, Resistenzbildung, geringe Breitbandwirkung), sodass Bedarf für neue Therapieoptionen besteht. Ziel des Projekts war daher die Entwicklung von anti-herpesviralen Breitbandinhibitoren mit reduzierter Resistenzentwicklung. Dieses Anforderungsprofil sollte unter anderem durch sogenannte dual-selektive niedermolekulare Inhibitoren, die gleichzeitig eine für den Virus wichtige Wirtskinase sowie eine Viruskinase inhibieren, realisiert werden.
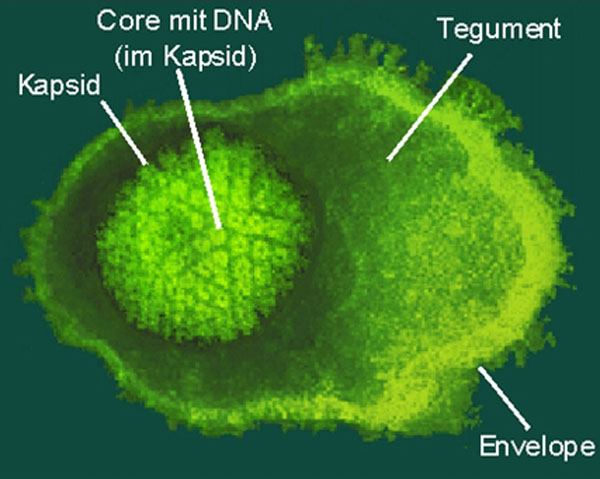
rechts: UL97-Kinase (Cartoondarstellung): computergeneriertes Homologiemodell (Quelle: 4SC AG)
Im Rahmen des Projekts wurden sowohl optimierte Proteinkinase-Inhibitoren mit starker antiviraler Aktivität als auch dual-selektive Proteinkinase-Inhibitoren mit einer besonders effizienten anti-herpesviralen Aktivität bearbeitet. Dabei wurden leistungsstarke Technologien für die Inhibitor-Optimierung weiterentwickelt und ein präklinischer Entwicklungskandidat nominiert. Als Targets validiert wurden die virale Proteinkinase pUL97 sowie mehrere humane Kinasen. Es konnten Hit-Substanzen für diese Kinasen gefunden und eine breite anti-herpesvirale Wirksamkeit für einige Inhibitoren gezeigt werden. Über die Kinaffinity-Matrix-Technologie wurden bisher wenig beachtete zelluläre Proteinkinasen als weitere vielversprechende Targets im Kontext der Herpesvirus-Infektion identifiziert. Die Untersuchung des Wirkmechanismus in Zellkulturmodellen zeigte, dass Kinaseinhibitoren meist in der frühen Virusreplikationsphase hemmen. Ein Nicht-Proteinkinase-Target (Dehydroorotat-Dehydrogenase) wurde zudem mittels einer In-vivo-Analyse im Mausmodell anhand der Hit-Substanz Cmp1 als sehr effektiv für antivirale Therapiemaßnahmen bestätigt.